| New in PolyA_DB v4 (Septemper 2025) |
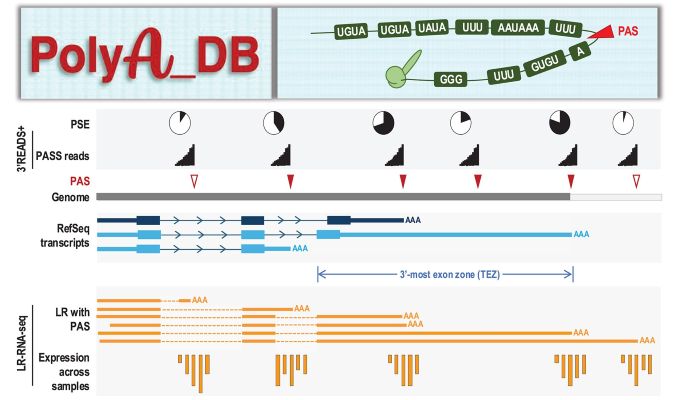
| PAS in v4 are exclusively identified by using 3′READS+ sequencing data. |
| V4 uses more data sets: 364 human and 451 mouse samples are included, totaling ~2.3 billion PAS-supporting reads for each species. |
| Long-read sequencing data (766.2 million reads from 227 human samples and 165 mouse samples) is now incorporated to validate PAS and substantially improve gene annotation, particularly for PAS located in 3′UTR extended regions and nested gene regions.. |
| PAS motifs are identified in the ±75 nt region surrounding the PAS, including UGUA, UAUA/AUAU, and UUUU in the upstream region, and UGUG/GUGU, GGGG, and UUUU in the downstream region. |
| PAS strength is predicted by the deep learning model-based tool PolyaStrength (Stroup EK & Ji Z. (2023) Nat Commun. 14:7378) and the scores are normalizaed to percentiles based on all PAS in the genome.
|